材料に潜む「欠陥」を予測する新AI技術を開発
- 新材料探索の新たな基盤技術として期待 -
2025/12/12
発表のポイント
- 実材料で重要な役割を担う「欠陥」のエネルギーを、人工知能(AI)で予測する手法を提案し、酸素空孔において世界最高の予測精度を実現しました。
- 開発したAIモデルを用いて有望な32種類のp型酸化物を提案しました。
- 本手法により、エネルギー変換デバイスや次世代エレクトロニクス材料の効率的な探索が期待されます。
概要
物質中に必ず存在する点欠陥(注1)は、材料特性を大きく左右する重要な要素であり、新材料の発見においてその特性の把握は不可欠です。しかし、欠陥形成エネルギーの評価には多大な計算資源が求められる量子力学計算(注2)が必要であり、実用材料探索の大きな障壁となっていました。
東北大学大学院工学研究科の澁井千紗大学院生(研究当時)、同大学金属材料研究所の清原 慎講師、Soungmin Bae助教、熊谷 悠教授らは、結晶をグラフとして扱うグラフニューラルネットワーク(GNN)(注3)を活用し、半導体や絶縁体中の欠陥がとり得る複数の電荷状態の形成エネルギーを単一モデルで同時に予測できる新しい枠組みを構築しました。これをもとに、酸素空孔の形成エネルギーを予測するAIモデルを構築し、世界最高精度の予測精度を実現しました。さらにこのモデルを用いておよそ2,000種類の酸化物材料を評価し、BaGaSbO をはじめとする有望な32種類のp型酸化物を提案しました。本成果は、これまで計算負荷の大きさから困難だった「欠陥を含む現実の材料の高速スクリーニング」を可能にするものであり、AI を活用した次世代材料開発の基盤技術として大きく貢献することが期待されます。
本研究成果は、2025年12月11日(現地時間)付で物理学分野を代表する国際学術誌Physical Review Letters にオンライン掲載されました。
研究の背景
近年、量子力学計算と機械学習の融合により、計算機を用いて新材料の発見に繋げる試みが産学問わず盛んに推進されていますが、その多くは「欠陥のない理想結晶」を前提としています。欠陥は実材料において、電気的・光学的・機械的特性などで重要な役割を担っていることは広く知られていますが、その欠陥形成エネルギーを量子力学計算で評価するには、膨大な計算資源を必要とします。こうした計算コストの高さが優れた実材料の発見を抑制してきました。
今回の取り組み
研究グループは人工知能(AI)を活用して、結晶構造の情報のみから非金属中の欠陥形成エネルギーを予測する新たな方法を開発しました。本手法では、グラフニューラルネットワーク(GNN)と呼ばれる結晶構造をグラフで表現する技術を用いることで、高精度に欠陥形成エネルギーを推定します。とくに、中性および正に帯電した酸素空孔など、電荷の異なる欠陥をひとつのAIモデルでまとめて扱えるようにした点が特徴です。従来は、電荷状態ごとに計算とモデル構築を個別に行う必要がありましたが、本研究では電荷の違いを共通の物差しで表現し、その情報を結晶構造とあわせてAIに学習させることで、多様な欠陥状態のエネルギーを一括して予測できるようにしました。例えば、本研究で対象とした酸素空孔は、非金属酸化物中では空孔サイトに残る電子数に応じて、中性、1価、2価の3つの価数を取り得ます。これらの電荷情報を適切に扱えるようにした結果、構造情報のみを入力とする汎用欠陥予測モデルとして、世界最高水準の精度を実現しました(図1)。これにより、従来は数日を要していた欠陥形成エネルギー計算を、瞬時に算出できるようになります。
本研究ではさらに、このAIモデルを用いて、およそ2,000種類の酸化物材料を対象に、p型半導体材料(注4)の探索を行いました。まず、酸化物のp型化を阻害する主因である酸素空孔によるホール補償が起きにくい酸化物をAIモデルでスクリーニングし、その後、有害元素および希少元素を含む酸化物を除外しました(図2(a))。さらに、量子力学計算により光吸収係数とホール有効質量を算出し、可視光をよく吸収して太陽電池用光吸収体として有望な材料や、高い透明性とp型伝導が期待できる透明導電酸化物として有望な材料を見いだしました(図2(b))。
本研究により、欠陥を含む「現実の材料」をAIで高速に予測・設計する道が開かれました。
今後の展開
本研究で提案した手法を基盤として、より幅広い結晶構造や多様な化学組成をもつ物質を対象に、任意の欠陥形成エネルギーを高精度に予測できる汎用AIモデルの開発に努めます。さらに、このモデルを応用して、点欠陥特性が鍵となるエネルギー変換デバイスや次世代エレクトロニクスの材料開発を効率的に進めるための基盤技術として発展させていきます。AIによる欠陥予測を材料設計の初期段階に組み込むことで、試行錯誤回数を減らした効率的な研究開発スタイルを実現し、持続可能なエネルギー社会の実現に貢献することを目指します。
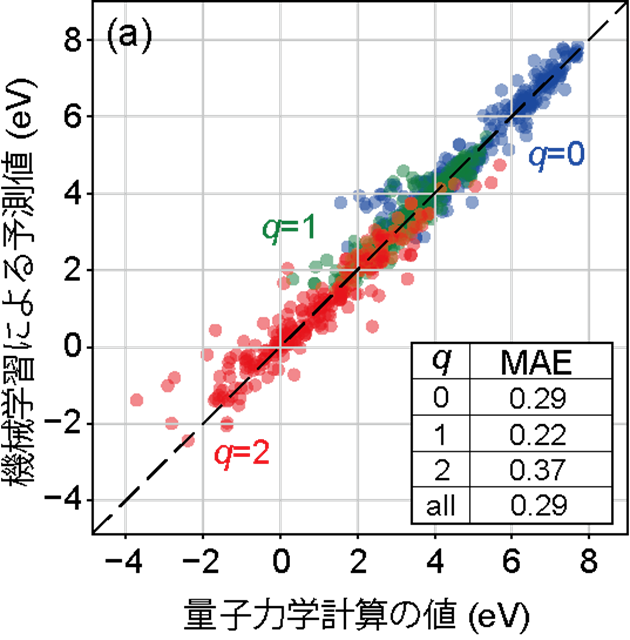
図1. 900酸化物中の酸素空孔形成エネルギー予測値で、横軸は、量子力学計算による値で、縦軸は機械学習による予測値を示しています。右下には、各電荷とそれらを合わせた際の平均絶対誤差をeVの単位で示しています。
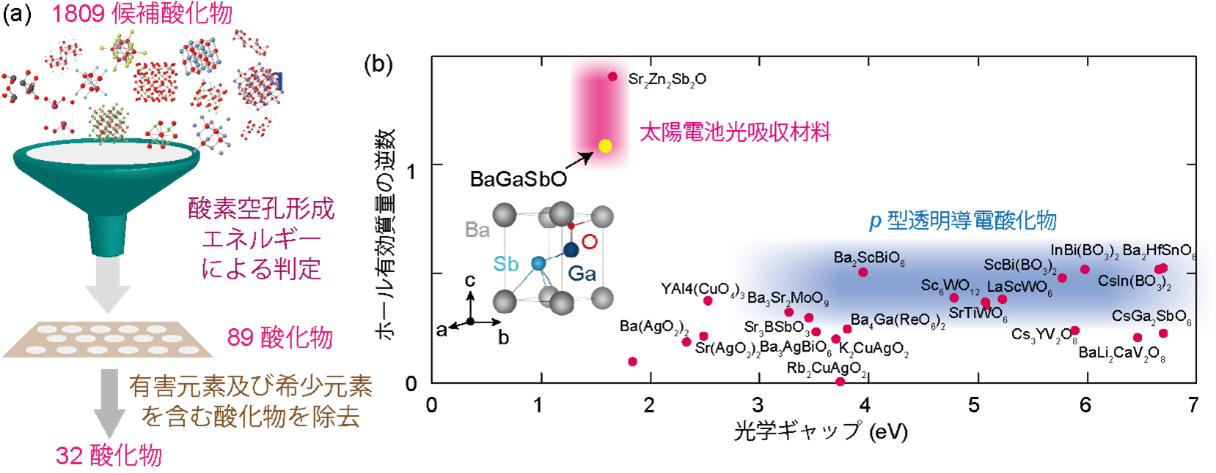
図2. (a)本研究におけるp型酸化物の仮想スクリーニングの概略図。(b)スクリーニングした32種の酸化物について、横軸に光学ギャップを、縦軸に自由電子質量を単位とした平均ホール有効質量の逆数をプロットしたものを示しています。また太陽電池の光吸収材料および透明導電性酸化物として有望な酸化物候補をそれぞれピンク色と青色でハイライトしています。
謝辞
本研究は、JSPS科研費基盤研究 B(JP22H01755、JP25K01486)、JST創発的研究支援事業(JPMJFR235S)、東北大学金属材料研究所先端エネルギー材料理工共創研究センター(E-IMR)の支援を受けて実施しました。また、掲載論文は『東北大学2025年度オープンアクセス推進のためのAPC支援事業』の支援を受けOpen Accessとなっています。(DOI:10.1103/h66h-y5k6)
用語説明
(注1)点欠陥
結晶中で原子が本来あるべき位置から欠ける(空孔)、別の原子に置き換わる(置換欠陥)、余分な原子が入り込む(格子間欠陥)などによって生じる、原子スケールの局所的な乱れである。ごく微量であっても電気伝導、光吸収、ドーピングの可否、材料の安定性などの物性を大きく左右するため、材料の性能を決定する重要因子となる。
(注2)量子力学計算
固体中の電子の振る舞いを量子力学に基づいて求める計算手法。特に固体材料では密度汎関数理論(DFT)が広く使われ、材料の安定性、電子構造、バンドギャップ、点欠陥形成エネルギーなどを原理的に評価できる。精度が高い一方で、計算コストが大きく、大規模材料探索には時間と計算資源が必要となる。
(注3)グラフニューラルネットワーク(GNN)
物質や分子などを「グラフ(点と線のつながり)」として扱い、その構造情報を学習する人工知能モデル。原子を“点(ノード)”、原子間の結合や距離を“線(エッジ)”として入力し、構造の特徴を自動的に抽出できるため、結晶構造を扱う材料科学の分野で広く利用されている。
(注4)p型半導体
電気を運ぶ主な担い手が「正孔(ホール)」と呼ばれる正の電荷をもつキャリアになっている半導体のこと。p型半導体は、n型半導体(電子が主なキャリアの半導体)と組み合わせることで、太陽電池やトランジスタなど多くの電子デバイスの基本構造を作るために不可欠である。p型化を実現する上で、欠陥などが電子を生み出してホールを打ち消してしまう「電荷補償」が起きないことが重要となるが、酸化物では酸素空孔がホール補償を引き起こしやすく、p型化が難しいことが知られている。
論文情報
著者: Shin Kiyohara, Chisa Shibui, Soungmin Bae and Yu Kumagai*
*責任著者: 東北大学金属材料研究所 教授 熊谷 悠
掲載誌: Physical Review Letters
DOI: 10.1103/h66h-y5k6
