高誘電体を高精度に予測・発見するAI手法を開発
―分解統合型AIが切り拓く高誘電体探索―
2026/04/08
発表のポイント
- 高誘電体で重要となる誘電率(注1)のイオンによる寄与を、結晶構造から高精度に予測する新しいAI手法を開発しました。
- 具体的には、イオン寄与を物理式に則り構成要素に分解し、それぞれを異なる機械学習モデルで予測して再構築する「分解統合型AI」により、世界最高精度を達成しました。
- 開発したAIモデルを用いて 、8,717 種類の酸化物を網羅的に探索し、31 種類の高誘電材料を新たに発見しました。それらのうち実験で誘電率が報告されている材料については、AI 予測値と実測値がいずれも高誘電率で一致し、モデルの信頼性を実証しました。
概要
物質の微視的構造が示す振る舞いを高精度に理解することは新規材料の創出や特性制御に不可欠ですが、その本質的理解には膨大な計算資源を要する量子力学計算(注2)が必要であり、大規模材料探索や迅速な設計の大きな障壁でした。
東北大学大学院工学研究科の滝川敦之大学院生、同大学金属材料研究所の清原慎講師、熊谷悠教授らのグループは、物質の原子配列情報のみから誘電率へのイオンの寄与を高精度に予測できるAI手法を開発しました。具体的には、物質の結晶構造をグラフとして表現し、グラフニューラルネットワーク(GNN)(注3)を用いてBorn有効電荷(注4)を予測するとともに、最新の機械学習ポテンシャルを活用してフォノン特性(注5)を算出し、それらを物理法則に基づく理論式により統合し、イオン寄与を求める新たな計算枠組みを構築しました。本手法により、従来は高精度な予測が困難であったイオン寄与を、量子力学計算と比べて大幅に少ない計算コストで高精度に予測できることを実証しました。さらに、本モデルにより数千種類の化合物を高速にスクリーニングし、LiBi4Nb3O14をはじめとする有望な新規材料候補群を抽出しました。本成果は、AIと物理理論を融合した次世代材料開発の基盤技術として重要な意義を持ち、材料探索の効率化と革新的材料の創出加速に貢献することが期待されます。
本研究成果は、2026年4月7日(現地時間)付で物理学分野の代表的な国際学術誌Physical Review Xにオンライン掲載されました。
研究の背景
半導体デバイスのさらなる微小化と高性能化を実現するためには、高誘電率と広いバンドギャップを兼ね備えた高誘電体材料の開発が不可欠です。しかし、その物性を高精度に評価するために用いられる量子力学計算は計算コストが非常に高く、評価可能な物質数が限られるという課題がありました。一方、近年、AIを活用した材料探索が注目を集めていますが、高誘電体において誘電率の大部分を占めるイオン寄与(εion)は予測が特に難しく、従来の機械学習手法では十分な精度を達成できていませんでした。このことが、高誘電体材料の大規模スクリーニング実現に向けた大きな障壁となっていました。
今回の取り組み
研究グループは、人工知能(AI)を活用し、結晶構造情報のみから誘電率のイオン寄与εionを高精度に予測する新たな手法を開発しました。本手法では、εion を直接予測するのではなく、Born有効電荷とフォノン特性という二つの基礎的物理量をそれぞれ機械学習により高精度に予測し、それらを第一原理に基づく解析式へ組み込むことで最終的なεionを再構築します。この「分解統合型」機械学習アプローチにより、従来の end-to-end 型モデルでは困難であった高誘電率領域での外挿性能を大幅に向上させ、誘電率予測の信頼性を飛躍的に高めることに成功しました。
具体的には、まずグラフニューラルネットワーク(GNN)を用いて Born 有効電荷を予測し、次に高精度機械学習ポテンシャル SevenNet によりフォノン特性を推定します。これらの予測結果を誘電率の理論式に代入することで、予測精度の指標である決定係数 R² は 0.91 を達成し、世界最高水準の精度を実現しました。特に、高誘電率材料という外挿領域においても安定した予測が可能であることを明らかにしました。
さらに、本モデルを用いて 8,717 種類の酸化物を対象とする網羅的スクリーニングを実施し、データベース上で誘電率が未計算であった 31 種類の高誘電体材料を新たに見出しました。そのうち、既に実験値が報告されている材料については、本手法の予測値と実測値がいずれも高誘電率で一致しており、モデルの高い信頼性が実証されました。
今後の展開
本手法により、今回対象とした酸化物にとどまらず、窒化物やハロゲン化物など他の物質群へも適用範囲を拡大していく予定です。また、本研究で新たに見出した有望な高誘電体材料については、今後、実験による誘電特性の精密測定やデバイス応用評価を進めることで、実用化に向けた研究の加速が期待されます。
さらに、本研究で提案した分解統合型機械学習アプローチは、誘電率に限らず、複数の基礎物理量の組み合わせによって決定される物性に対して広く適用可能な汎用的手法です。本成果は、AIと物理理論を融合することで高い外挿性能を実現する新たな枠組みを提示するものであり、AIを活用した材料探索に新しいパラダイムをもたらすことが期待されます。
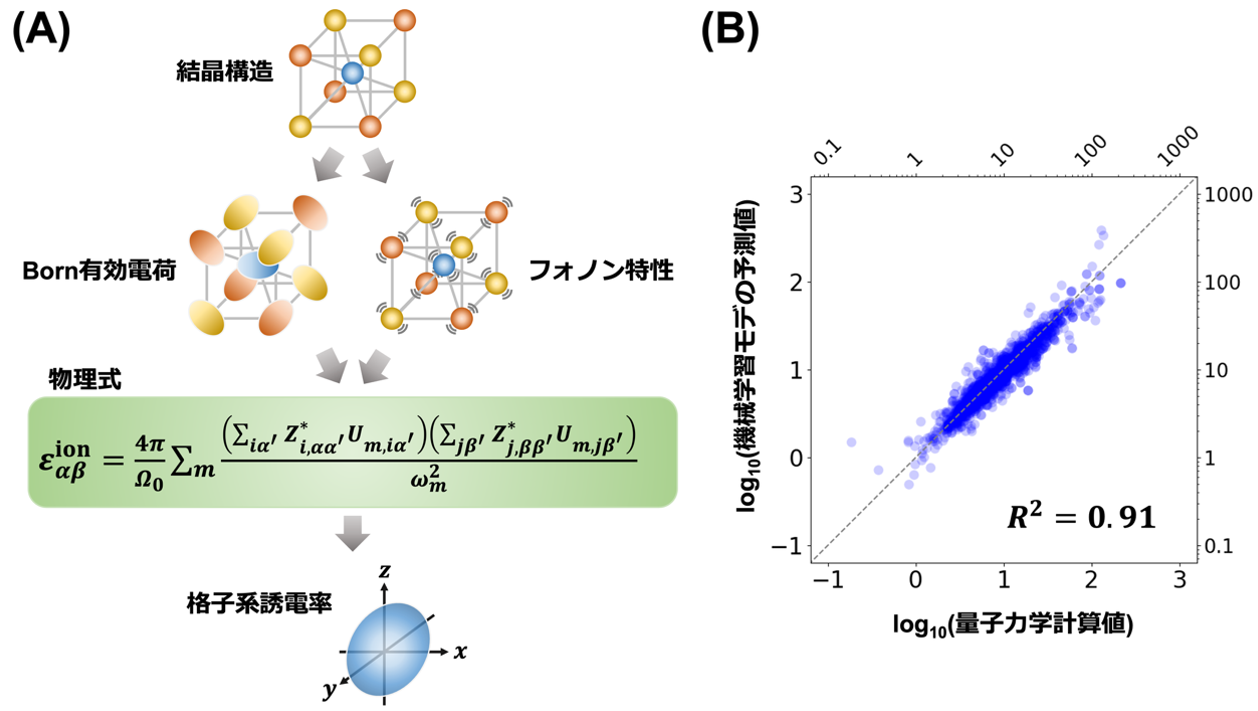
図1. (A)本研究において構築した機械学習を用いた予測手法の概略図。結晶構造の情報からBorn有効電荷とフォノン特性を個別の機械学習モデルで予測し、図に示す物理式を用いて誘電率のイオン寄与を再構築します。(B)928酸化物中のイオン寄与予測値で、横軸は量子力学計算による値を、縦軸は機械学習による予測値を示しています。R2は決定係数という予測精度を表す指標で、1に近いほど予測精度が高いことを示します。
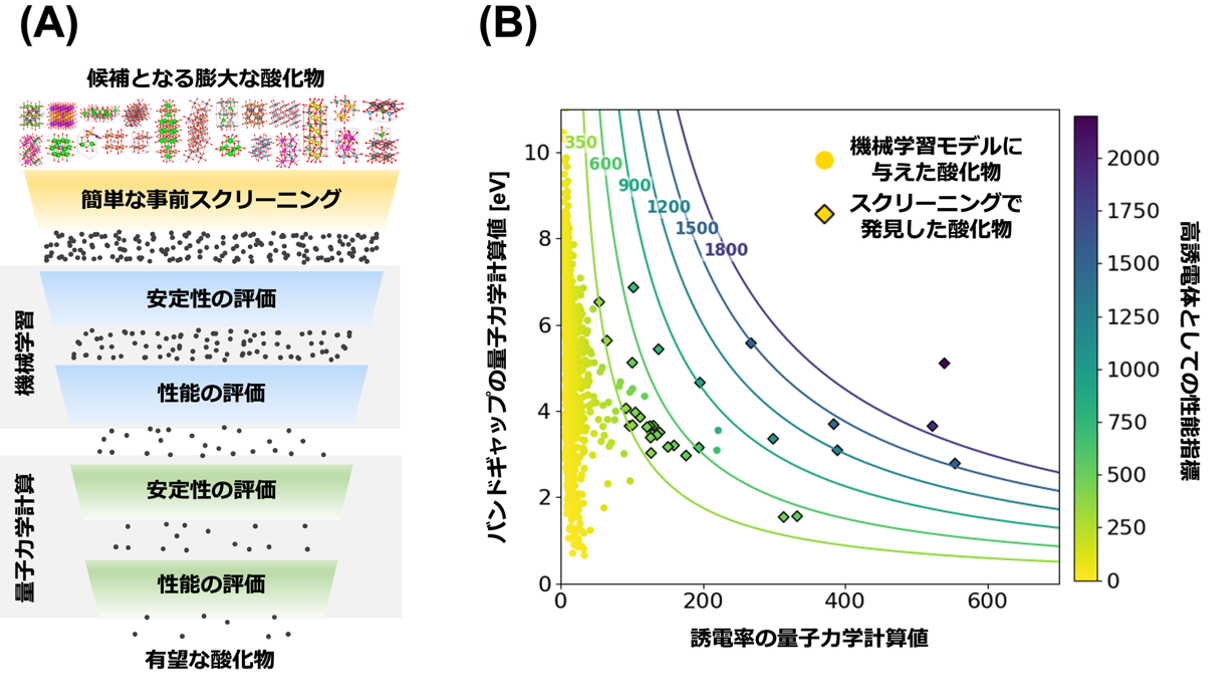
図2. (A)本研究における高誘電酸化物探索の概略図。(B)選抜した31種の酸化物について、横軸を量子力学計算により得られた誘電率とバンドギャップ、縦軸をバンドギャップの量子力学計算値としてプロットしたものを♢で示しています。色は、高誘電体の性能指標で、バンドギャップと誘電率の積で表現され、この値が高いほど有望な高誘電酸化物であることを示唆しています。参考として、本研究で機械学習モデルを訓練するために与えた酸化物を○で示しています。
謝辞
本研究は、JSPS科研費基盤研究 B(JP22H01755、JP22H01755)、JST創発的研究支援事業(JPMJFR235S)、旭硝子財団、東北大学金属材料研究所先端エネルギー材料理工共創研究センター(E-IMR)の支援を受けて実施しました。また、掲載論文は『東北大学2025年度オープンアクセス推進のためのAPC支援事業』の支援を受けOpen Accessとなっています。
用語説明
(注1)誘電率
物質が電場を受けたときにどれだけ電場を遮蔽できるかを示す物理量で、値が大きいほどより強く電場を遮蔽することができます。固体では、電場により電子やイオンがわずかに移動して分極が生じ、その応答の大きさが誘電率として表されます。
(注2)量子力学計算
固体中の電子の振る舞いを量子力学に基づいて求める計算手法。特に固体材料では密度汎関数理論(DFT)が広く使われ、材料の安定性、電子構造、バンドギャップ、点欠陥形成エネルギーなどを原理的に評価できます。精度が高い一方で、計算コストが大きく、大規模材料探索には時間と計算資源が必要となります。
(注3)グラフニューラルネットワーク(GNN)
物質や分子などを「グラフ(点と線のつながり)」として扱い、その構造情報を学習する人工知能モデル。原子を“点(ノード)”、原子間の結合や距離を“線(エッジ)”として入力し、構造の特徴を自動的に抽出できるため、結晶構造を扱う材料科学の分野で広く利用されています。
(注4)Born有効電荷
結晶中の原子が電場を受けて変位したときに、どれだけ強く電気分極を生み出すかを表す物理量です。単なる形式的なイオンの電荷とは異なり、原子の周囲の電子状態の変化も含めた実効的な電荷を示します。この値が大きいほど、原子の振動が誘電率に強く寄与するため、Born有効電荷は高誘電体材料におけるイオン寄与を決定する重要な指標の一つです。
(注5)フォノン特性
結晶中の原子振動の性質を表すものです。原子は格子点の周りで振動しており、その集団的な振動を量子力学的に扱った概念がフォノンです。振動の周波数やモードは、特に誘電率のイオン寄与を決める重要な要素となります。
論文情報
著者: Atsushi Takigawa, Shin Kiyohara*, and Yu Kumagai*
*責任著者: 東北大学金属材料研究所 講師 清原 慎、教授 熊谷 悠
掲載誌: Physical Review X
DOI: 10.1103/28wr-w896
